- 14 Dec, 2025 *
Before We Talk About AI, We Need to Talk About Why Proteins Are Ridiculously Complicated
You know what’s wild? Right now, as you’re reading this, there are approximately 20,000 different types of proteins working inside your body. Not 20,000 total proteins, 20,000 TYPES. The actual number of protein molecules? Billions. Trillions if we’re counting across all your cells.
Each one has a specific job. Each one has a specific shape. And if even ONE type folds wrong, one could get Alzheimer’s, cystic fibrosis, sickle cell anemia, Parkinson’s, Huntington’s, mad cow disease, or any of thousands of other diseases collectively called "protein misfolding diseases."
Your body makes these proteins perfectly, billions of times a day, in every single one of your 37 tril…
- 14 Dec, 2025 *
Before We Talk About AI, We Need to Talk About Why Proteins Are Ridiculously Complicated
You know what’s wild? Right now, as you’re reading this, there are approximately 20,000 different types of proteins working inside your body. Not 20,000 total proteins, 20,000 TYPES. The actual number of protein molecules? Billions. Trillions if we’re counting across all your cells.
Each one has a specific job. Each one has a specific shape. And if even ONE type folds wrong, one could get Alzheimer’s, cystic fibrosis, sickle cell anemia, Parkinson’s, Huntington’s, mad cow disease, or any of thousands of other diseases collectively called "protein misfolding diseases."
Your body makes these proteins perfectly, billions of times a day, in every single one of your 37 trillion cells, without asking your opinion, without requiring a user manual, without ever attending a protein folding workshop.
Scientists spent 50 years, FIFTY YEARS, trying to figure out how to PREDICT what shape a protein would fold into based on its amino acid sequence. Entire careers were built on this problem. Nobel Prizes were awarded for incremental progress. Supercomputers were dedicated to simulating single protein folds that took weeks to complete.
Then AI companies showed up in 2020 and said "we got this" and solved it in an afternoon.
And now? Now we’re not just predicting shapes, we’re DESIGNING entirely new proteins that have never existed in nature. Proteins that can break down plastic. Proteins that can capture carbon dioxide. Proteins that can target cancer cells with sniper precision. We’re playing God with molecules and it’s working.
But before I tell you how NVIDIA went from making GPUs that render explosions in Call of Duty to designing molecules that might cure cancer, you need to understand what proteins actually are and why this problem was so stupidly, impossibly, hilariously hard that it became one of biology’s grand challenges alongside "how does consciousness work" and "what is dark matter."
Let’s start from scratch. Forget everything you learned in high school biology. We’re doing this right.
Proteins 101: The LEGO Bricks of Life (Except Way More Complicated and They Build Themselves)
Remember from my previous articles: your DNA gets transcribed into RNA, which gets translated into proteins. That’s the central dogma. DNA → RNA → Protein. Information flows one way. (Mostly. Retroviruses are weird. Don’t worry about it.)
But what IS a protein? And I mean really, fundamentally, at the molecular level?
A protein is a chain of amino acids that folds into a specific 3D shape, and that shape determines what the protein does.
That’s it. That’s the entire definition. A chain. That folds. Into a shape. That does stuff.
But as with literally everything in biology, the devil is in the details. And the details are where things get interesting (and by interesting, I mean "ridiculously complicated but in a cool way")
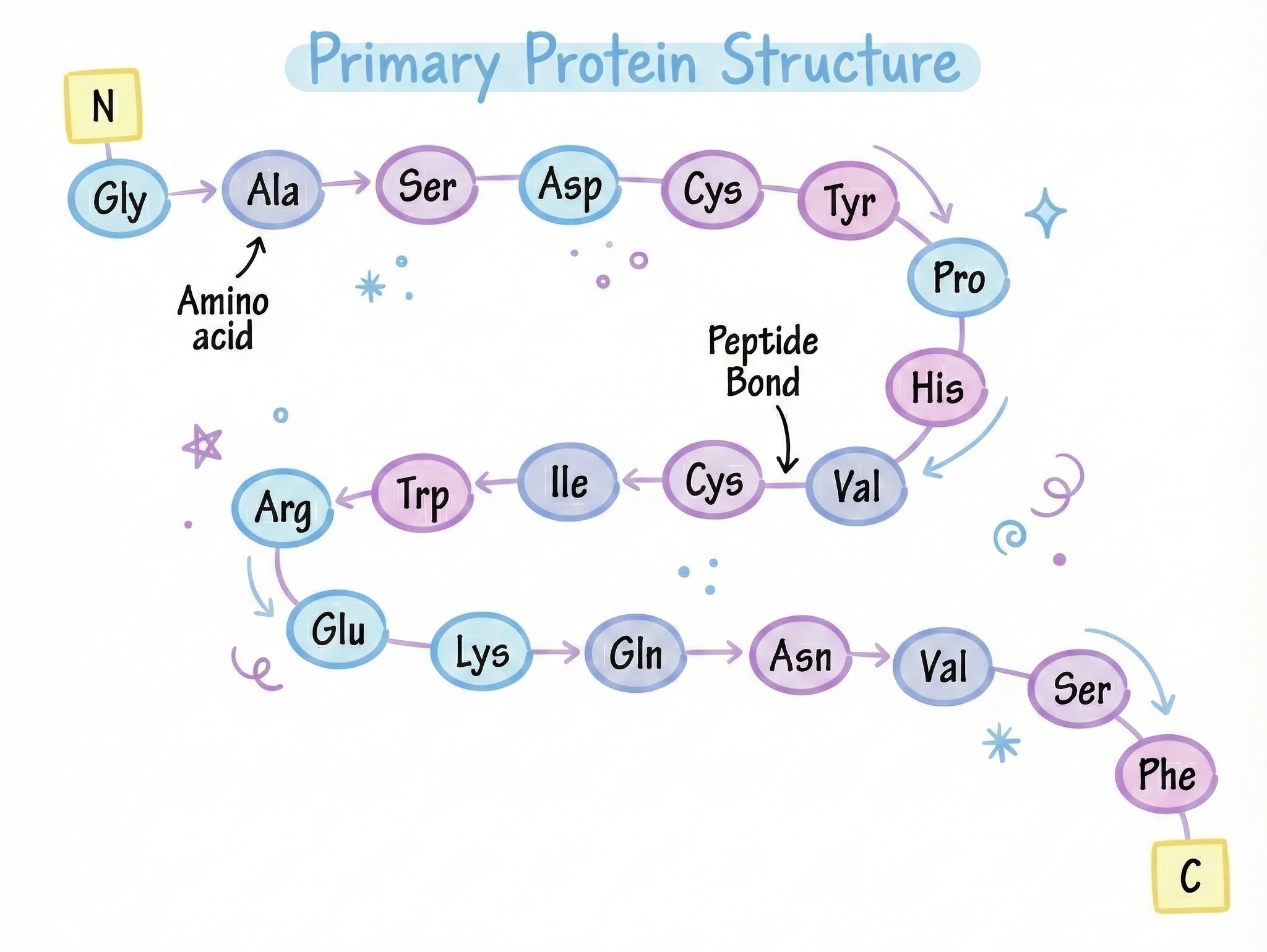
Amino Acids: The 20-Letter Alphabet That Writes Every Function in Your Body
There are 20 standard amino acids that your body uses to build proteins. (There are actually a few more non-standard ones, but let’s not complicate things yet.) Think of them as letters in an alphabet. But instead of making words and sentences, they make functional machines.
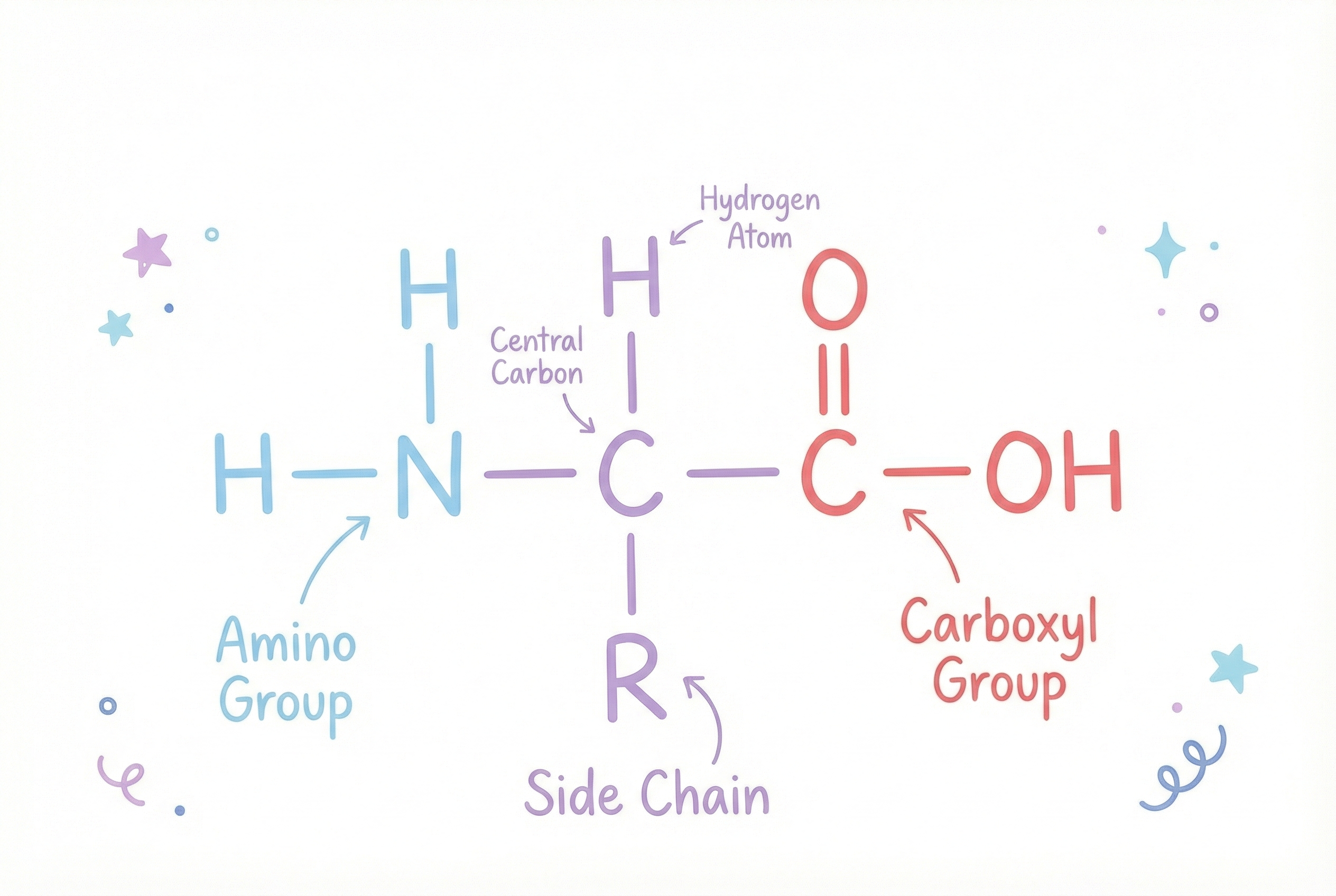
Each amino acid has the same basic structure:
- An amino group (NH₂) on one end, this is the "amino" in amino acid
- A carboxyl group (COOH) on the other end, this is the "acid" part
- A hydrogen atom attached to the central carbon
- A unique side chain (called an R group) attached to the central carbon That side chain, the R group, is where all the personality lives. It’s what makes each amino acid unique.
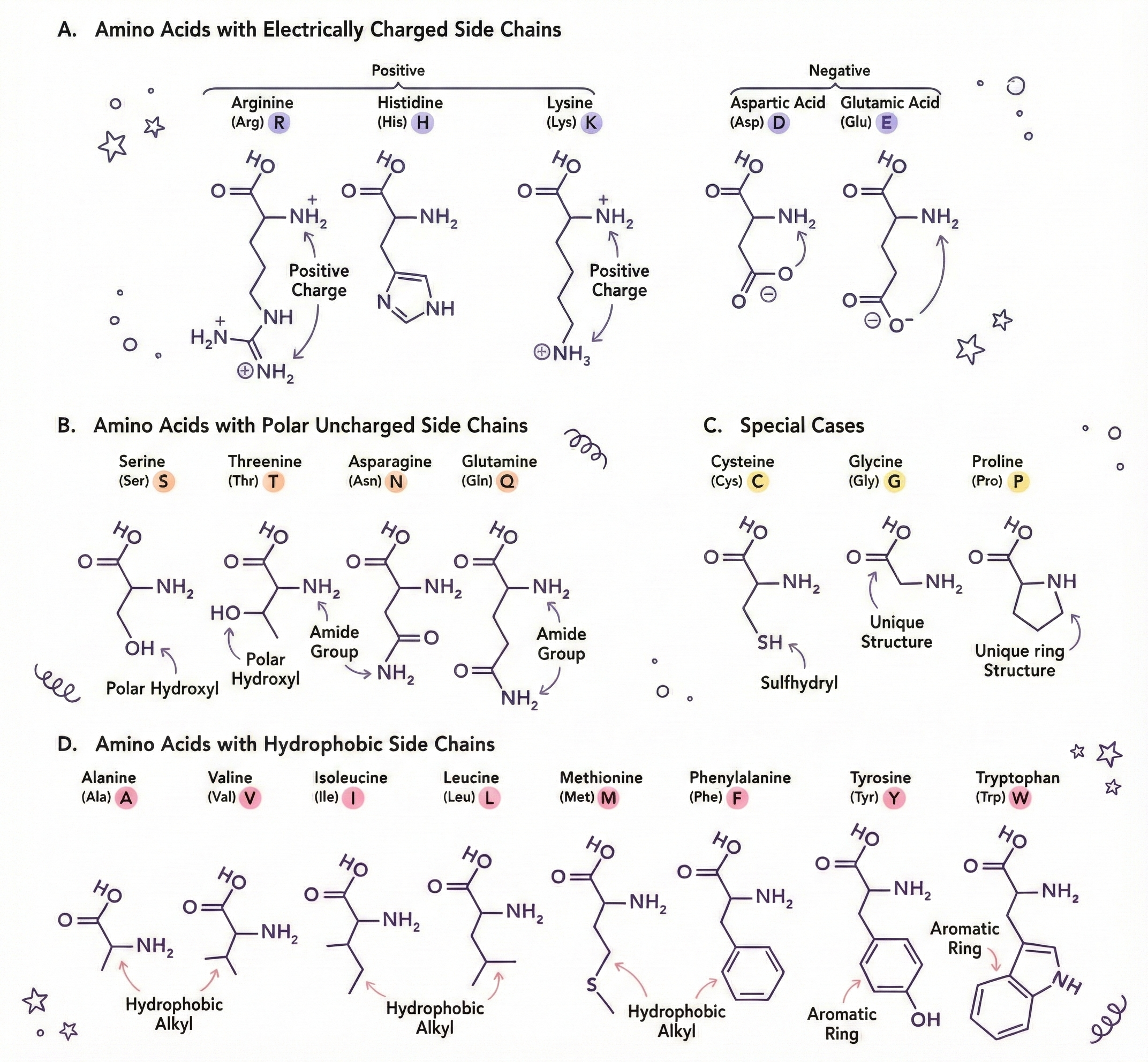
Let Me Introduce You to Some Amino Acids (They Have Personalities)
- Glycine: The smallest amino acid. Its side chain is just a single hydrogen atom. It’s tiny, flexible, fits anywhere. The perfect team player. Gets along with everyone.
- Proline: The rebel. Has a ring structure that creates kinks in protein chains. Shows up and makes everything bend around it. Doesn’t conform to the rules. We respect the energy.
- Cysteine: Contains sulfur. Two cysteines can form a disulfide bond (S-S) with each other, creating a chemical "staple" that holds parts of proteins together. It’s the duct tape of amino acids.
- Tryptophan: Huge, bulky, takes up space. Often found buried in protein cores because it’s hydrophobic (hates water). The introvert of amino acids.
- Aspartic acid and Glutamic acid: Negatively charged. Basically walk around with a permanent bad attitude, repelling other negatively charged amino acids and attracting positive ones.
- Lysine and Arginine: Positively charged. The optimists. Attract negativity (literally). Create electrostatic interactions that stabilize proteins.
- Phenylalanine, Leucine, Isoleucine, Valine: Hydrophobic. HATE water. In an aqueous environment, they huddle together in the protein’s core like people at a party who only know each other.
The point is: these 20 amino acids can be arranged in ANY order and in ANY length to create proteins. And your body picks the exact right order to make each protein work.
The side chains determine:
- Whether the amino acid likes or hates water
- Whether it’s charged (positive, negative, or neutral)
- How big it is (size matters for packing efficiency)
- Whether it’s rigid or flexible
- What chemical reactions it can participate in
The Combinatorial Explosion of Possibilities
A typical protein has 200-400 amino acids. Some have thousands. Titin, the largest known protein in humans, has 34,350 amino acids. It’s literally a molecular spring that provides elasticity to muscle tissue.
Let’s do some math that will hurt your brain:
For a protein that’s just 100 amino acids long, there are 20^100 possible sequences. That’s 1.27 × 10^130 possible combinations.
For reference:
- There are about 10^80 atoms in the observable universe
- There are about 10^24 stars in the observable universe
- The number of possible 100-amino-acid sequences is 10^50 times MORE than the number of atoms in the universe
And most of those sequences? They don’t fold into anything useful. They’re junk. They aggregate into clumps. They get degraded by cellular quality control. Only a TINY fraction of possible sequences fold into stable, functional proteins.
Nature had to search this impossibly vast space and find the sequences that actually work. And it did this through random mutation and natural selection over 3.5 billion years. Evolution is the ultimate brute-force search algorithm.
But we don’t have 3.5 billion years. We want to design proteins NOW.
Folding, The Part Where the Magic Happens (And Also Where Everything Can Go Wrong)
When a ribosome finishes making a protein (remember translation from my last article?), it spits out a long, floppy, completely linear chain of amino acids. This chain is called a polypeptide, literally "many peptides" because each amino acid is connected to the next by a peptide bond.
The peptide bond forms between the carboxyl group of one amino acid and the amino group of the next:
...—NH—CHR—CO—NH—CHR—CO—NH—CHR—CO—...
This creates a backbone (the repeating NH-CHR-CO pattern) with side chains (R groups) sticking out.
And then, immediately, while the ribosome is still finishing the rest of the chain, something magical happens:
The chain starts folding itself.
No chaperone proteins initially (those come later if needed). No instructions. No assembly manual. No quality control inspector. The amino acids just start interacting with each other based on their chemical properties, and the whole thing spontaneously collapses into a compact, functional 3D structure.
This is called spontaneous folding or self-assembly, and it’s one of the most beautiful phenomena in molecular biology.
The Forces That Drive Folding
Protein folding is driven by thermodynamics, specifically, the search for the lowest free energy state (most stable configuration). Multiple forces contribute:
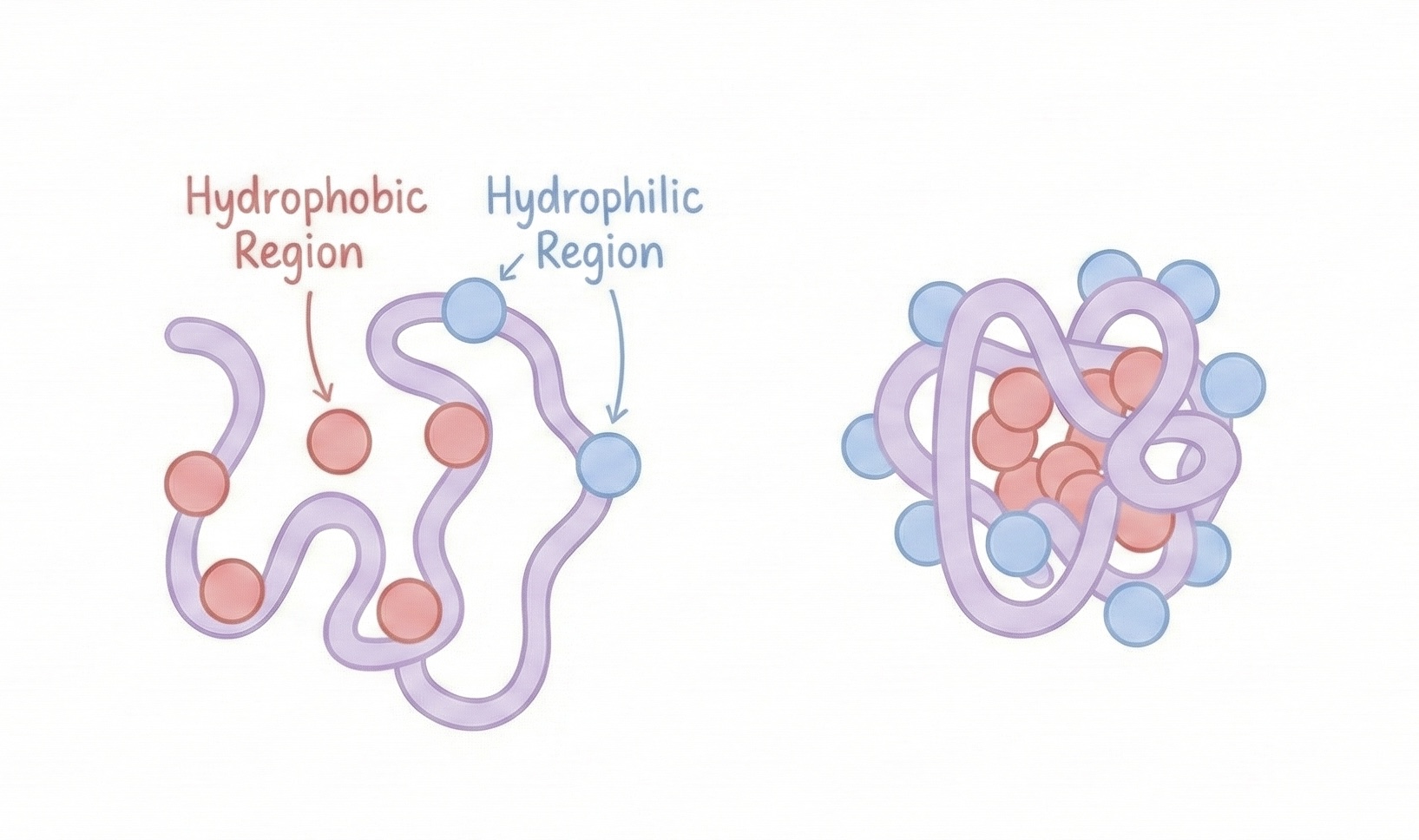
1. The Hydrophobic Effect (The Big One)
This is the primary driving force for most proteins. Hydrophobic amino acids (like leucine, valine, phenylalanine) are energetically unfavorable in water. Water molecules have to organize around them in structured "cages," which decreases entropy (disorder).
The system wants to maximize entropy. So what happens? Hydrophobic amino acids cluster together in the protein’s core, away from water. This releases the ordered water molecules back into the bulk solution, increasing overall entropy.
Meanwhile, hydrophilic amino acids (charged and polar ones) stay on the surface, happily interacting with water.
This creates a protein structure with:
- A hydrophobic core (oil-like interior)
- A hydrophilic shell (water-loving exterior)
Like a molecular M&M. Except instead of chocolate, it’s biochemistry.
2. Hydrogen Bonds (The Backbone of Structure)
Hydrogen bonds form between:
- The carbonyl oxygen (C=O) and amide hydrogen (N-H) of the backbone → creates secondary structures
- Side chains with hydroxyl (-OH), amine (-NH₂), or carboxyl (-COOH) groups
Individually, hydrogen bonds are weak (about 5% the strength of a covalent bond). But proteins have HUNDREDS of them. Collectively, they’re incredibly strong.
They’re responsible for:
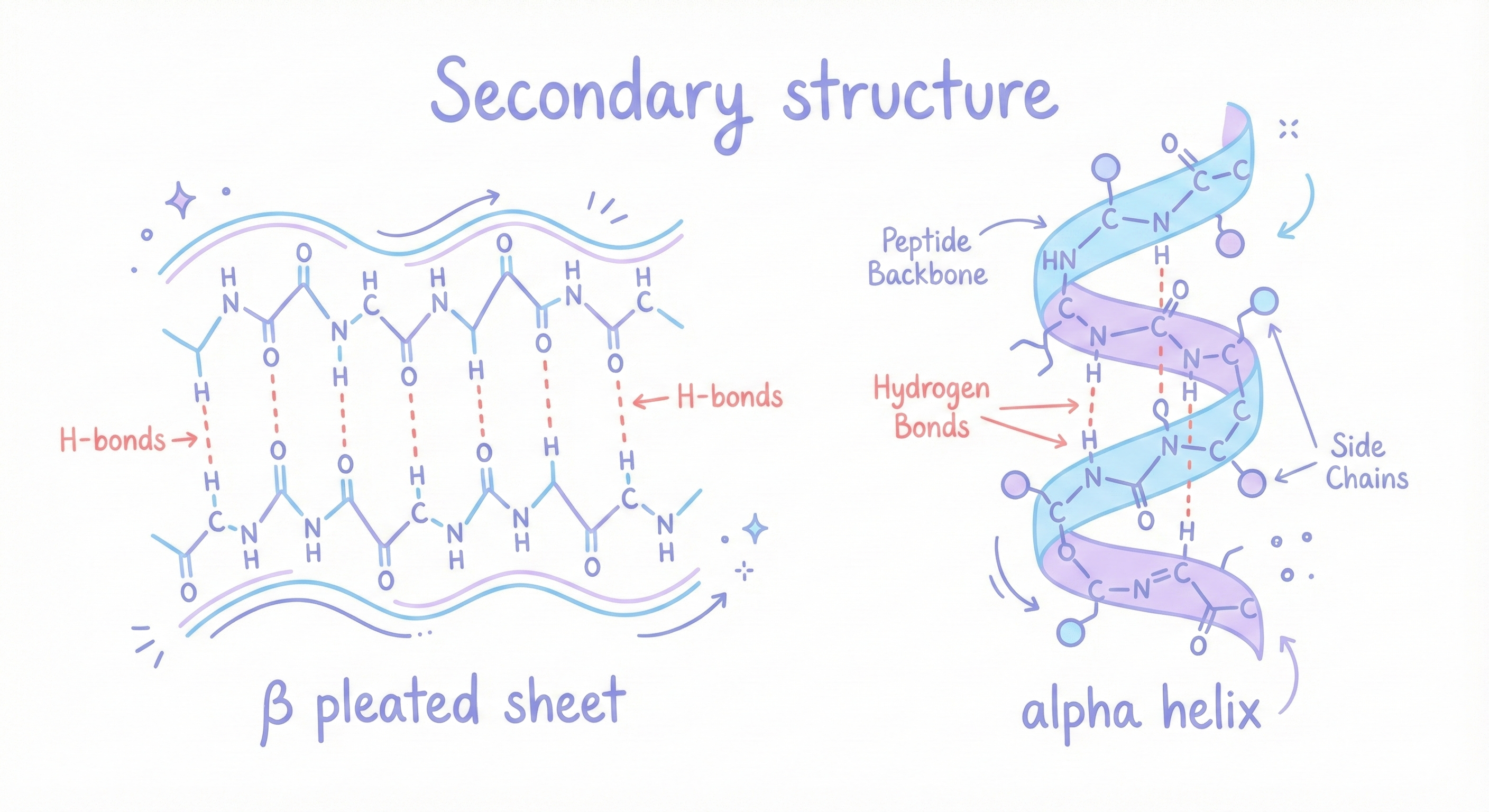
- Alpha helices: The backbone coils into a spiral, with hydrogen bonds between the C=O of residue n and the N-H of residue n+4. It’s like a molecular spring. Very stable. Common in proteins.
- Beta sheets: The backbone extends into a zigzag sheet, with hydrogen bonds between parallel or antiparallel strands. It’s like a pleated paper fan. Also very stable. Also very common.
These are called secondary structures, local patterns in the protein backbone.
3. Electrostatic Interactions (Salt Bridges)
Oppositely charged amino acids attract each other:
- Lysine (+) attracts Aspartate (-)
- Arginine (+) attracts Glutamate (-)
These are called salt bridges or ion pairs. They’re strong and help stabilize the folded structure.
4. Disulfide Bonds (The Chemical Staples)
When two cysteine residues come close together, their sulfur atoms can form a disulfide bond (S-S). This is a COVALENT bond, much stronger than the other interactions.
Disulfide bonds are like staples that hold parts of the protein together. They’re especially common in:
- Extracellular proteins (outside cells, where the environment is oxidizing)
- Antibodies (which need to be stable in harsh environments)
Inside cells (reducing environment), disulfide bonds are rare.
5. Van der Waals Forces (The Weak but Numerous)
When atoms get very close, they experience weak attractive forces called van der Waals interactions. They’re tiny individually, but proteins have THOUSANDS of atoms in close contact, so collectively they matter.
6. Entropy (The Desire for Disorder)
Folding DECREASES entropy (the protein goes from a floppy, disordered chain to a compact, ordered structure). This is thermodynamically unfavorable. But remember: folding releases water molecules from around hydrophobic residues, which INCREASES entropy. The net effect? Folding is favorable overall.
The Folding Timeline: Milliseconds to Seconds
How fast does this happen?
Small proteins (50-100 amino acids) can fold in microseconds to milliseconds. Larger proteins take seconds.
For reference:
- Millisecond = 10^-3 seconds
- Microsecond = 10^-6 seconds
Your cells are making proteins and folding them CONSTANTLY. Every second. Right now. While you read this.
And here’s the crazy part: the folded structure is reproducible. Given the same sequence, you get the same structure. Every time. It’s deterministic (mostly, there are exceptions called intrinsically disordered proteins, but let’s not go there).
This means the folding information is ENCODED in the amino acid sequence. The sequence contains all the instructions needed to fold into the correct shape. But humans don’t know how to READ those instructions directly. We can see the sequence. We can see the final structure. But predicting one from the other? That took 50 years to figure out.
The Final Product: The Native Structure
The final 3D shape is called the protein’s native structure. It has several levels of organization:
Primary structure: The linear sequence of amino acids. Just the order.
Secondary structure: Local patterns (alpha helices and beta sheets).
Tertiary structure: The full 3D arrangement of the entire protein chain.
Quaternary structure: If multiple protein chains come together (like hemoglobin, which has 4 chains), how they’re arranged relative to each other.
The native structure is the functional form. This is the shape that DOES the biology.
And this is where things get critical.
Shape = Function (And This Is Why Protein Folding Is Life or Death)
Here’s the most important concept in all of protein biology, and I cannot stress this enough:
A protein’s function is ENTIRELY determined by its 3D shape.
Not the amino acid sequence. Not the chemical properties of individual residues. The three-dimensional structure.
Change the shape even slightly, and the protein stops working. Change it drastically, and you get disease.
Let me give you examples that show just how insanely specific this is.
Example 1: Enzymes and the Lock-and-Key Model
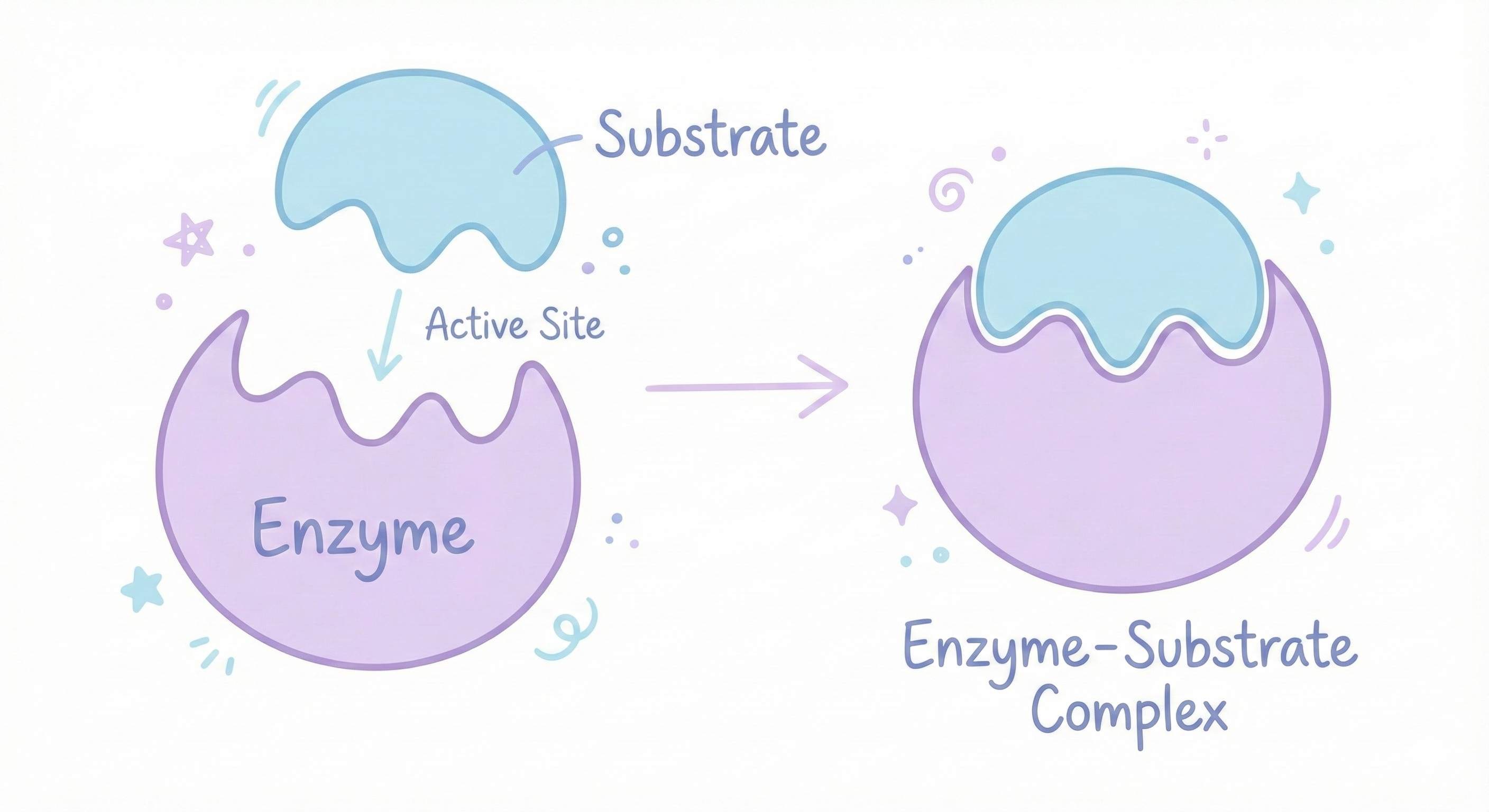
Enzymes are proteins that speed up chemical reactions. Without them, most biological reactions would happen so slowly that you’d be dead. Your cells would be frozen in chemical slow-motion. Enzymes have a specific pocket called an active site, a precisely shaped cavity where the chemical reaction happens. The substrate (the molecule the enzyme works on) fits into this pocket like a key in a lock. The fit is SPECIFIC. If the shape is even slightly wrong, the substrate won’t fit. The reaction won’t happen. The enzyme is useless.
Lactase (The Enzyme That Digests Milk Sugar)
Lactase is the enzyme that breaks down lactose (milk sugar). If you’re lactose intolerant, it’s because your body either stopped making lactase or makes a misfolded version that doesn’t work. Result? Lactose sits in your intestines. Gut bacteria ferment it. You get gas, bloating, diarrhea. One misfolded protein = digestive chaos. You can’t drink milk because your protein has the wrong shape. That’s how specific this is.
Example 2: Antibodies and Immune Recognition
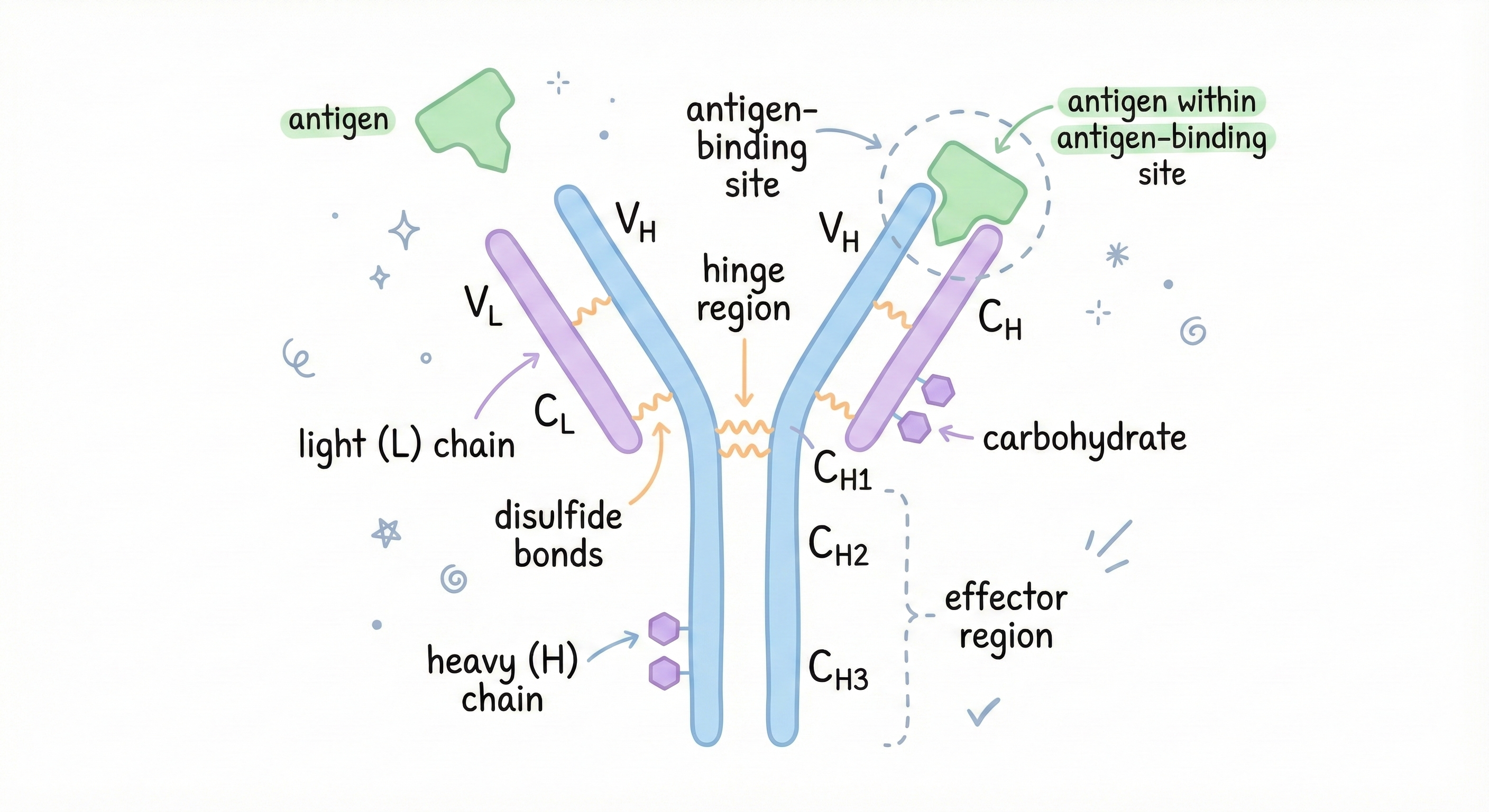
Your immune system has to recognize millions of different threats: viruses, bacteria, toxins, parasites. It does this with antibodies, Y-shaped proteins that bind to specific invaders. Each antibody is custom-shaped to recognize a specific molecular pattern (called an antigen) on the surface of an invader. The tips of the Y are shaped to fit that specific target. The fit is so precise that an antibody designed for the flu virus won’t recognize the common cold virus. Different shapes = different antibodies needed.
This is why vaccines work. You expose your immune system to a harmless version of a pathogen, your body makes antibodies with the right shape to recognize it, and now you’re protected. If the antibody shape is wrong, your immune system won’t recognize the threat. You get sick.
Modern medicine exploits this by designing custom antibodies as drugs:
- Herceptin: Treats breast cancer by binding to specific receptors on cancer cells
- Humira: Treats autoimmune diseases by blocking inflammatory proteins
- Keytruda: Unleashes your immune system to attack cancer cells
These are literally designer proteins with custom shapes targeting specific molecules. Billion-dollar drugs that work because the shape is right.
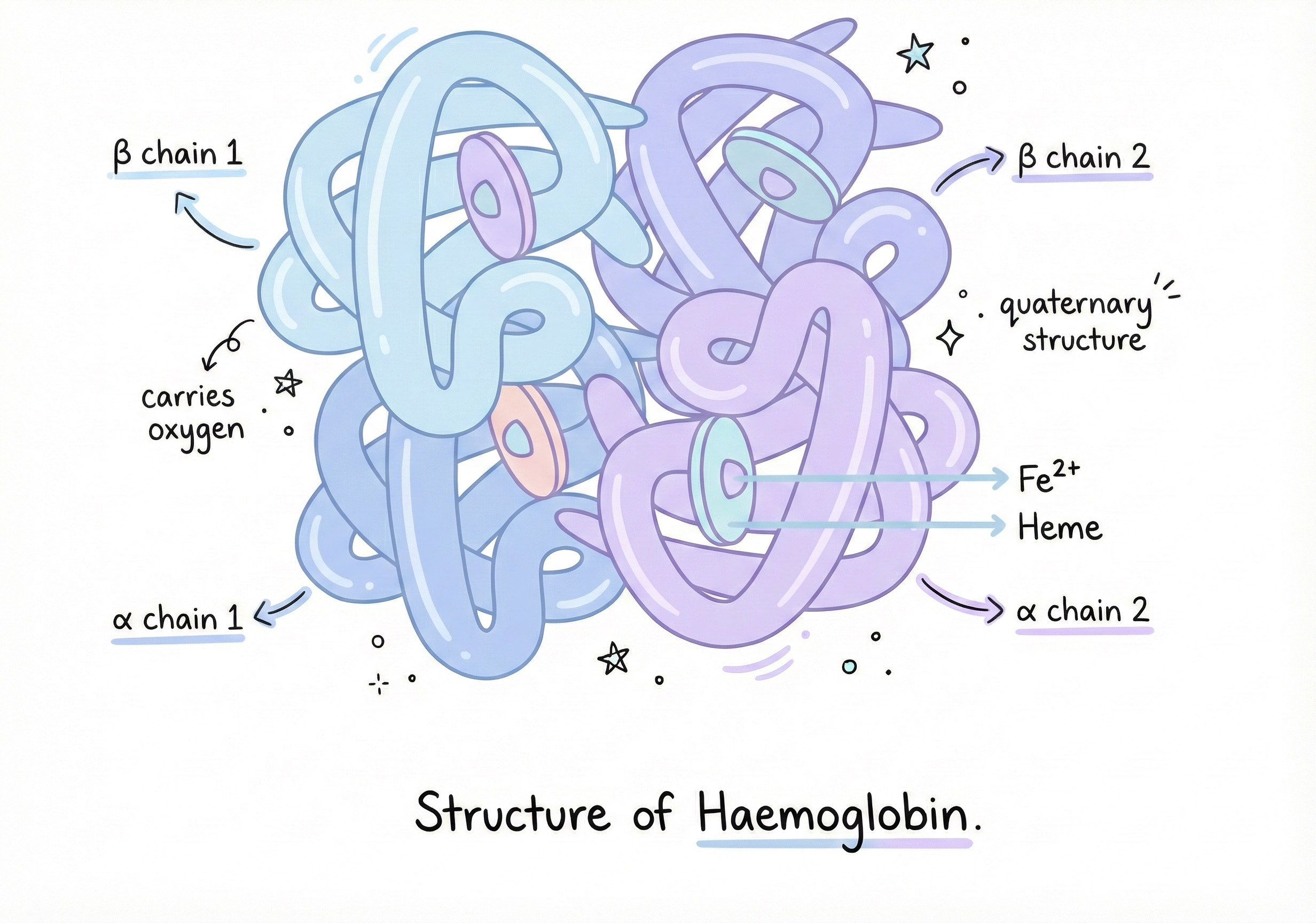
Example 3: Haemoglobin and the Oxygen Transport System
Haemoglobin carries oxygen in your blood. It’s shaped like a four-leaf clover with pockets that hold iron atoms, which bind oxygen. The shape is critical for function. Haemoglobin picks up oxygen in your lungs (where O₂ is abundant) and releases it in your tissues (where O₂ is scarce). But if you change just ONE amino acid... Sickle Cell Anemia is caused by a single mutation:
Position 6 in the beta chain of haemoglobin Glutamic acid (charged, hydrophilic) → Valine (hydrophobic)
That’s it. One letter out of 146 amino acids in that chain.
But valine is hydrophobic. It creates a sticky patch on the surface of the haemoglobin molecule. When haemoglobin releases oxygen, this patch is exposed. Hydrophobic patches love to stick together. So sickle haemoglobin molecules clump together, forming long fibers. These fibers deform red blood cells into sickle (crescent) shapes. Sickled cells:
- Get stuck in blood vessels → pain, organ damage
- Break apart easily → anemia
- Don’t carry oxygen well
One amino acid. One shape change. Lifelong disease.
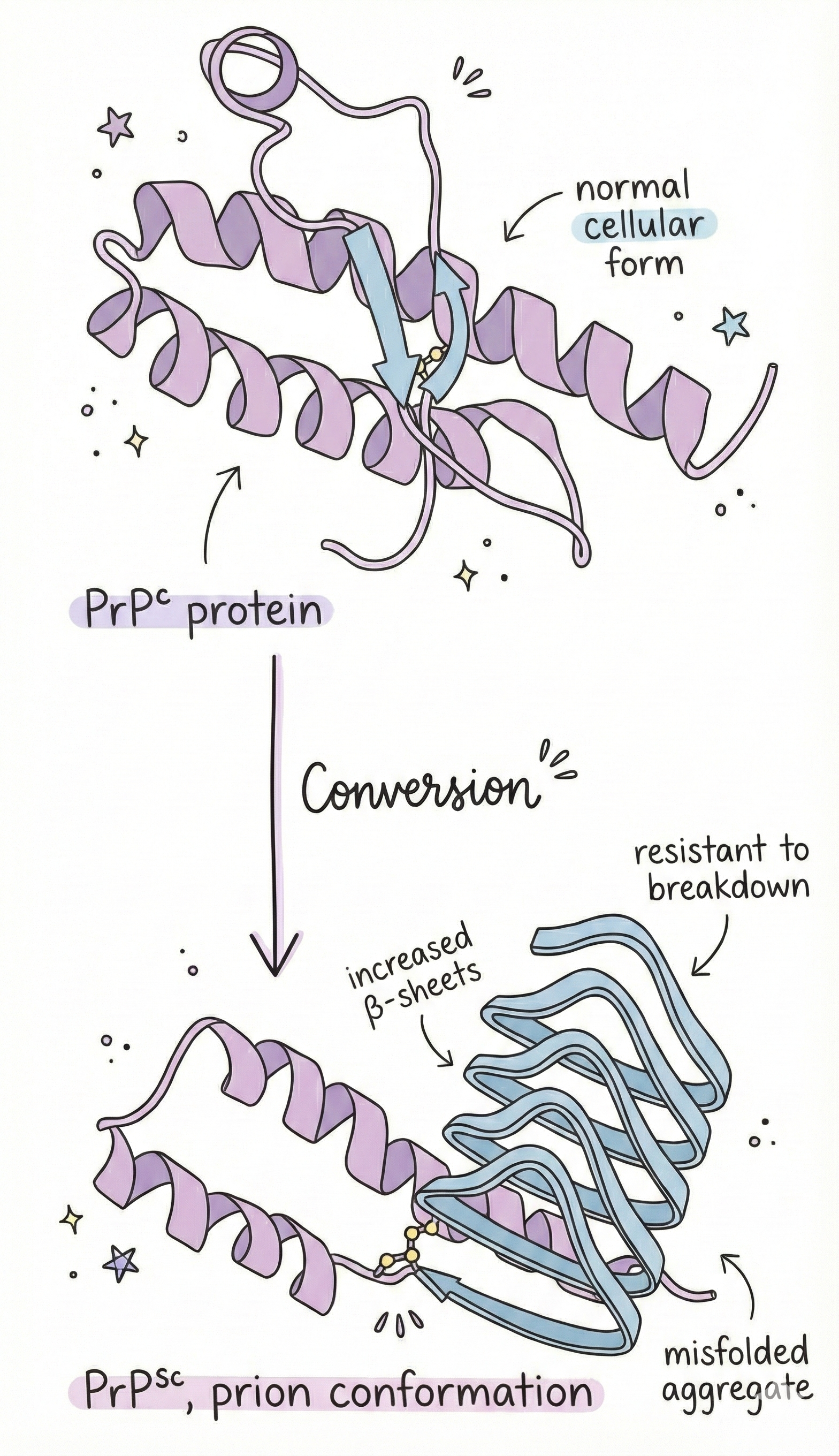
Example 4: Prions and the Horror of Misfolding
Here’s where it gets truly terrifying. Prions are misfolded proteins that can convert normal proteins into the misfolded form, spreading like an infection. They cause diseases like:
- Mad cow disease
- Creutzfeldt-Jakob disease
- Kuru
- Fatal familial insomnia
The protein involved is called PrP (prion protein). Everyone has it. It’s a normal protein on the surface of neurons. But PrP can misfold into a different shape, same amino acid sequence, different structure. This misfolded version (PrP^Sc) is:
- Protease-resistant (can’t be broken down)
- Forms aggregates (clumps together)
- Converts normal PrP into the misfolded form
It’s autocatalytic. Self-replicating. And it destroys brain tissue.
One can get prion diseases by:
- Eating infected tissue (mad cow)
- Inheriting a mutation that makes PrP more likely to misfold (genetic)
- Spontaneous misfolding (sporadic, rare but it happens)
There’s no cure. It’s 100% fatal. And it’s all because of protein shape.
The Levinthal Paradox: Why Protein Folding Should Be Impossible (But Isn’t)
In 1969, a scientist named Cyrus Levinthal did some math and realized something disturbing: Protein folding shouldn’t work.
The Math That Breaks Reality
Consider a protein with 100 amino acids. Each amino acid has bonds that can rotate, and each bond has maybe 3 stable angles. So there are roughly 10^95 possible shapes the protein could adopt. Now, let’s say the protein can try one shape every picosecond (10^-12 seconds). That’s incredibly fast, molecular vibrations happen on that timescale. How long would it take to try all possible shapes to find the correct one? 10^83 seconds. The universe is about 10^17 seconds old. 10^83 seconds is 10^66 times longer than the age of the universe. If proteins had to randomly search for the correct fold, it would take longer than the universe has existed. But proteins fold in milliseconds. This is the Levinthal Paradox. Folding should be impossible. But it happens. Every time.
The Answer: Proteins Don’t Search Randomly
Proteins follow a folding pathway, they don’t try every possible shape. They take shortcuts.
Think of it like this:
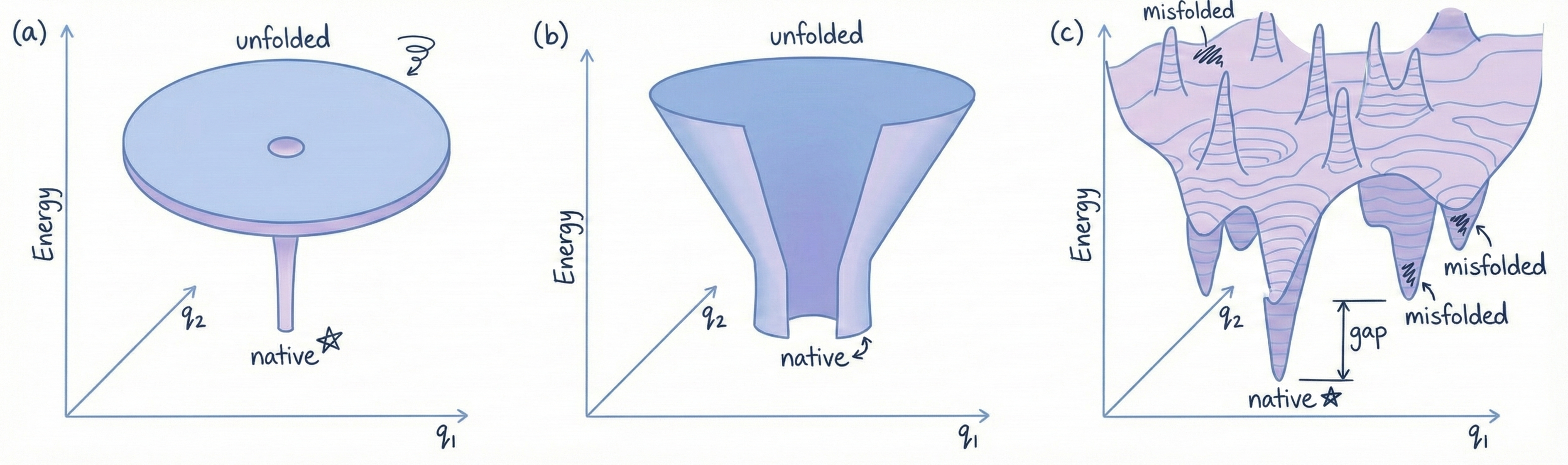
Imagine a landscape with hills and valleys. The native structure is the deepest valley (lowest energy state). If the protein randomly wandered around, it would take forever to find the valley.
But the landscape is shaped like a funnel:
- The protein starts at the top (unfolded, high energy)
- Local interactions form (hydrophobic collapse, alpha helices, beta strands)
- These constrain the possible conformations, the protein is now halfway down the funnel
- More interactions form, further stabilizing the structure
- The protein slides down the funnel toward the native state
It’s guided by the energy landscape encoded in the amino acid sequence. Evolution figured out sequences that fold efficiently.
Nature is smarter than random searching. Who knew.
The Protein Folding Problem: A 50-Year Quest
So, proteins fold into specific shapes based on their sequences. Great.
Here’s what scientists wanted to do:
Give me an amino acid sequence (like: MKTAYIAKQRQISFVKSHF...) and I’ll tell you what 3D shape it will fold into.
Simple request. Insanely hard problem.
This is called the protein folding problem, and it’s been one of the grand challenges of biology since the 1960s.
Why Scientists Cared
If you can predict protein structure from sequence, you can:
- Understand diseases: Know which mutations cause misfolding
- Design drugs: Target specific pockets in disease-related proteins
- Engineer enzymes: Create custom proteins with desired functions
- Understand evolution: See how protein structures change over time
But we couldn’t do it. We tried for 50 years. And mostly failed.
Why It’s Ridiculously Hard
1. The Search Space is Incomprehensibly Vast
We already covered this. 10^95 possible conformations for a 100-amino-acid protein. Even with folding pathways, the space is enormous.
2. The Interactions Are Complicated
Every amino acid interacts with every other amino acid. For a 100-amino-acid protein, that’s nearly 5,000 possible pairwise interactions. And they all influence each other simultaneously. It’s like trying to solve a Rubik’s cube where every move affects every other square in unpredictable ways.
3. You Have to Model Water
Proteins fold in water. Water molecules interact with the protein, forming hydrogen bonds, pushing hydrophobic parts inward, stabilizing charged regions. You can’t model the protein in isolation. You need to simulate thousands of water molecules too. And water is WEIRD, its properties (hydrogen bonding, high dielectric constant) make it computationally expensive to model.
4. Small Changes Have Big Effects
Change one amino acid and the whole structure can change. It’s not a linear relationship. The folding landscape is rugged, small mutations can shift the entire energy funnel.
5. It’s a Physics Problem
Protein folding is governed by thermodynamics. You need to calculate the free energy of every possible conformation and find the global minimum (most stable state).
This requires simulating quantum mechanical interactions between thousands of atoms. Computationally, it’s a nightmare.
Early Attempts
Scientists tried everything:
X-ray Crystallography (1950s onward):
Grow protein crystals → Blast them with X-rays → X-rays diffract off the atoms → Analyze the diffraction pattern → Reconstruct the 3D structure
This WORKS, but:
- Takes months (growing crystals is hard)
- Requires pure protein samples
- Only works for proteins that crystallize (many don’t)
- Gives you the structure of proteins you ALREADY HAVE, not predictions for new ones
NMR Spectroscopy (1980s onward):
Put protein in a magnetic field → Use radio waves to probe the positions of atoms → Reconstruct the structure from the data
This also works, but:
- Limited to small proteins (<30 kDa)
- Requires high concentrations
- Expensive and time-consuming
Cryo-Electron Microscopy (2010s):
Flash-freeze proteins → Image them with an electron microscope → Average thousands of images to get high resolution
This revolutionized structural biology (2017 Nobel Prize) but:
- Still expensive
- Still requires specialized equipment
- Still only gives you structures of existing proteins
None of these methods PREDICT structures. They DETERMINE structures experimentally.
Computational Approaches:
Scientists tried to simulate folding computationally.
Molecular Dynamics (MD) Simulations: Model every atom in the protein and surrounding water → Calculate forces between atoms using physics equations → Simulate the motion of atoms over time (Newton’s laws) → Watch the protein fold
Sounds great! Except:
- It’s computationally INSANE
- You need femtosecond timesteps (10^-15 seconds)
- Proteins fold in milliseconds (10^-3 seconds)
- That’s 10^12 timesteps to simulate one folding event
- For a medium-sized protein in water: ~100,000 atoms
- Modern supercomputers: days to weeks for one simulation
And even then, you might miss the correct fold or get trapped in a metastable state (local energy minimum that’s not the global minimum).
Rosetta (2000s):
A software developed by David Baker’s lab that uses:
- Energy functions (estimations of how stable a given conformation is)
- Monte Carlo sampling (randomly try different conformations, keep the good ones)
- Fragment assembly (use known protein fragments as building blocks)
Rosetta was better than nothing. It could sometimes predict structures for small proteins or proteins similar to known structures.
CASP: The Protein Folding Olympics
In 1994, researchers created CASP, Critical Assessment of Structure Prediction. Every two years, teams compete to predict protein structures. Organizers choose proteins whose structures are about to be solved experimentally, teams submit predictions, and then the real structures are revealed.
Scores range from 0 to 100. Above 90 is considered competitive with experimental accuracy. For 25 years, the best scores hovered around 40-60 for difficult targets. Progress was incremental. Slow. Frustrating.
AlphaFold 1: The Warning Shot (2018)
In CASP13 (2018), a team from DeepMind (Google’s AI lab) entered a protein structure prediction method called AlphaFold. It used deep learning, neural networks trained on known protein structures. AlphaFold 1 achieved a median GDT score of 58.9, placing first overall. The biology community took notice. This was the first time a machine learning approach significantly outperformed traditional methods. But it wasn’t revolutionary. It was good, not great. There were still errors. Difficult targets were still difficult. Researchers thought: "Okay, AI is promising, but we’re not there yet."
Then came 2020.
AlphaFold 2: The Moment Everything Changed (2020)
In November 2020, CASP14 results were announced.
DeepMind’s AlphaFold 2 achieved a median GDT score of 92.4.
Let me put this in perspective:
- Previous best: ~60
- AlphaFold 2: 92.4
- Scores above 90 are considered competitive with experimental methods
AlphaFold 2 essentially SOLVED the protein folding problem.
For 87% of targets, it achieved GDT > 90. For some targets, it was MORE accurate than the experimental structures (because X-ray crystallography has its own errors).
DeepMind open-sourced the code and released the AlphaFold Protein Structure Database, predicted structures for 200 million proteins (essentially every known protein sequence).
For free.
How AlphaFold Works
Proteins are like language. A protein sequence is a string of letters (amino acids). Those letters follow rules (chemistry). The structure is the "meaning" of the sequence. And modern AI is REALLY good at understanding language patterns. That’s what powers large language models. AlphaFold adapted the same technology, transformers with attention mechanisms, for proteins.
The Process:
- Input: Amino acid sequence
- Find related sequences: Search databases for similar proteins (evolution often conserves structure even when sequences change)
- Attention mechanism: A neural network learns relationships between amino acids, which ones are close in 3D space, which ones interact
- Structure prediction: Build the 3D coordinates based on learned patterns
- Output: Predicted structure with confidence scores
AlphaFold doesn’t simulate physics. It recognizes patterns learned from 170,000+ known protein structures. And it works. Ridiculously well.
But AlphaFold Wasn’t Perfect. AlphaFold 2 was groundbreaking, but gaps remained:
- Speed: Each prediction took hours. If you want to screen thousands of protein designs for drug discovery, that’s a problem.
- Design: AlphaFold predicts structure from sequence. It doesn’t design sequences for a desired structure. That’s harder.
- Novel proteins: For completely new folds (not similar to anything in training data), accuracy dropped.
- Dynamics: Some proteins don’t have a single fixed shape, they’re flexible. AlphaFold predicts one static structure. This is where NVIDIA enters the picture.
Why NVIDIA?
NVIDIA makes GPUs, your Graphics Processing Units. They were originally designed for rendering video game graphics. But GPUs are incredibly good at parallel processing, doing thousands of calculations simultaneously. And guess what needs massive parallel processing?
- Training neural networks
- Running protein predictions
- Simulating molecular interactions
- Screening thousands of drug candidates
NVIDIA realized: the same hardware that powers gaming can power drug discovery. So they didn’t just optimize AlphaFold to run faster on GPUs, they built an entire ecosystem for biological research:
- BioNeMo: Language models for proteins (think GPT, but for biology)
- ProteinDT: Design proteins by describing them in plain English
- La-Proteina: Generate entirely new proteins from scratch
- ESM models: Understand protein sequences like language
- OpenFold optimizations: Make structure prediction 138x faster
And they partnered with pharmaceutical giants, Pfizer, Amgen, AstraZeneca, who are using these tools to design drugs RIGHT NOW.
Next, we’ll cover:
- How NVIDIA made protein prediction 138x faster
- BioNeMo: GPT for biology (language models trained on proteins)
- ProteinDT: Designing proteins by describing them in English
- La-Proteina & Proteína: Generating new proteins that have never existed
- ESM models: Teaching AI to "read" protein sequences
- The complete pipeline: from "I want a drug" to "here’s a protein candidate"
- Real applications: companies using this NOW to cure diseases
- The future: personalized medicine, synthetic biology, designer organisms
Basically: how gaming GPUs became the most important tool in modern drug discovery. And why this matters even if you’ve never thought about proteins before.
Part 2 coming soon. This is where things get truly wild.
Disclaimer: Everything in this article is scientifically accurate. Proteins really fold in milliseconds. AlphaFold really solved a 50-year-old problem. Your gaming GPU really uses the same architecture as drug discovery platforms. Biology is weird, AI is powerful, and we’re living in the future.
#NVIDIA [#Protein folding](https://aval.bearblog.dev/blog/?q=Protein folding) [#Protein structure](https://aval.bearblog.dev/blog/?q=Protein structure)